基于多层次特征融合的药物-靶标亲和力预测方法及系统
本发明涉及药物-靶标亲和力预测领域,具体而言,涉及一种基于多层次特征融合的药物-靶标亲和力预测方法及系统。
背景技术:
1、药物和靶标之间的相互作用调控着众多生物过程,在生物体内在扮演着至关重要的角色。例如,药物分子与蛋白酶的活性位点结合,可以增强或抑制酶的活性,进而影响生物体内的新陈代谢。在信号转导过程中,药物与受体蛋白的相互作用能够调节细胞内外的信号传递,影响细胞的生长、分化和凋亡等生理功能。药物-靶标亲和力预测是利用计算方法预测药物-靶标之间的结合亲和力值。结合亲和力值是衡量药物与靶标相互作用强度的重要指标。亲和力值的大小直接反映了药物与靶标之间的结合强度,这有助于研究人员更加全面地对药物分子的活性进行评估,从而选择具有最佳亲和力值的候选化合物进行后续研究,因此药物-靶标亲和力预测研究对于药物发现和药物开发具有重要的意义。
2、亲和力色谱和蛋白质微阵列是两种常用的生物实验方法,用于测量药物与靶标之间的结合亲和力值。对于一个特定的靶标,药理学家在寻找安全且有效的靶向药物时,往往需要进行数千次的实验尝试,这一过程不仅耗时而且成本高昂。因此,计算预测方法因其高效和成本低廉的特点,引起了研究者们的极大兴趣。这些计算方法能够快速预测药物与靶标之间的结合亲和力,显著提高药物筛选的效率,有效降低研发成本,极大程度地缩短药物发现和药物开发进程。
3、根据输入数据的类型,现有的计算方法可以分为基于结构的和基于非结构的。分子对接和分子动力学模拟是两种比较著名的基于结构的预测方法,它们以化合物和蛋白质的三维结构作为输入。分子对接方法是根据几何匹配和能量匹配原则,使用评分函数来评估药物-靶标之间的相互作用强度,不仅能够识别配体的结合模式,还能提供与结合亲和力相关的定量对接评分。而分子动力学模拟的优势在于能够提供药物-靶标相互作用过程中能量变化和结构变化的详细信息,通过这些信息可以揭示药物和靶标之间的相互作用机制,从而指导新药物或新靶标的发现。然而,由于缺乏足够的化合物和蛋白质三维结构数据,这些方法的适用性受到严重限制;而且,这些方法计算成本高昂,而且耗时较长,整体效率低下。
4、基于非结构的方法是以序列作为输入,如kronrls和simboost。在kronrls方法中,核是衡量两个分子之间相似性的函数。kronrls使用内核的方法构建药物分子之间或靶标之间的相似性,然后使用正则化最小二乘回归(rls)框架预测亲和力值。相似性匹配方法simboost根据药物和靶标之间的相似性为每个药物、靶标和药物-靶标对构建特征,然后将基于相似性的特征用作梯度提升机的输入,从而预测药物-靶标亲和力值。这些方法的局限性在于忽略了每个分子中单个组分的详细特征,并且严重依赖相似性度量来预测绑定亲和力值,不可避免地会导致较大的偏差。
5、近年来,如平行合成和高通量筛选等新的实验技术显著增加了生物医学研究的现有数据量。基于非结构的方法使用深度学习技术来自动从大量数据中提取特征。在基于深度学习的方法中,输入数据的表示对模型的性能有重要的影响。deepdta模型采用药物序列和蛋白质序列作为输入,通过卷积神经网络(convolutional neural networks,cnn)捕获序列中的信息。然后,将获得的蛋白质和化合物的特征向量进行拼接,并输入到两个全连接层中预测结合亲和力。随后,hakime等人扩展了deepdta模型,命名为widedta。该方法首先将药物和蛋白质的序列推广为高阶特征。例如,药物通过最常见的亚结构(ligand maximumcommon substructures,lmcs)来表示,而蛋白质通过prosite的最保守亚序列(proteindomain profiles or motifs,pdm)来表示。zhao等人提出gansdta,利用生成对抗网络(generative adversarial networks,gan)学习标记和未标记序列中的信息,并使用卷积回归预测结合亲和力得分。zeng等人也利用了药物和蛋白质的序列表示,提出关系感知的自注意力模块对药物中原子之间的相对位置进行建模,考虑了原子之间的相关性,并建立多头注意力模块模拟药物-靶标对之间的相似性作为dta预测的交互信息。tsubaki等人提出将图神经网络(graph neural network,gnn)应用于药物-靶标亲和力预测。该模型将药物的化学结构表示为分子图(molecular graph),并使用gnn和cnn分别学习分子图和蛋白质序列的低维特征向量。gao等人的研究利用gnn来学习药物的分子图信息,并采用长短期记忆网络(lstm)来捕捉蛋白质序列的特征。nguyen等人提出了graphdta模型,该模型将药物表示为分子图,并利用图卷积神经网络(graph convolutional network,gcn)来提取特征向量。这些方法充分利用了药物和蛋白质的结构信息,通过深度学习模型对其进行建模,能够更好地捕捉它们之间的关联性和相互作用信息。这样的研究在药物-靶标相互作用预测领域具有重要的应用价值,为开发新药和药物重定位提供了有力的支持。同时,利用一维卷积神经网络获取蛋白质序列的特征向量,并将两个特征向量拼接后通过全连接层预测亲和力值。karim等人利用cnn和lstm的组合来描述化合物和蛋白质。当cnn应用于时间序列数据时,该方法提取高度信息化的特征以编码局部时间模式。为了捕捉全局时间模式,该模型将lstm层添加在卷积层之上。同时,该模型提出了一个双向注意力机制,用于编码蛋白质亚序列与化合物亚结构之间的相互作用,得到的注意力系数表示化合物和蛋白质之间的结合强度。lin等人提出了deepgs模型[49],该模型将蕴含药物拓扑信息的分子图表征与包含上下文信息的药物序列表征相结合,并利用卷积神经网络从蛋白质序列中获取蛋白质序列表征用于预测结合亲和力值。
6、但是,上述方法中主要存在以下问题:现有的预测dta方法没有将序列、分子图和分子指纹这三种表征有效结合起来表示。现有的方法只使用了单一或者最多两种特征来表示药物或蛋白质,不能有效全面地提取药物和蛋白质的多层次特征。现有的方法只使用蛋白质的序列特征,而没有考虑到蛋白质序列中氨基酸数量和类型的信息。
7、有鉴于此,特提出本发明。
技术实现思路
1、有鉴于此,本发明提出了一种基于多层次特征融合的dta预测方法,使用了分子序列、分子图、分子指纹来表示药物,使用蛋白质序列和n-grams特征来表示靶标。这种多层次的特征表示方法能够捕捉到药物和靶标之间复杂的相互作用模式,从而提高亲和力预测的精度,并且相较于早期预测药物与靶标的亲和力的方法,大大节省了研发成本,此外还具有增强药物设计的针对性,促进个性化医疗,支持药物重定位,提升药物安全性评估的效果。
2、具体地,本发明是通过以下技术方案实现的:
3、本发明提供了一种基于多层次特征融合的药物-靶标亲和力预测方法,包括如下步骤:
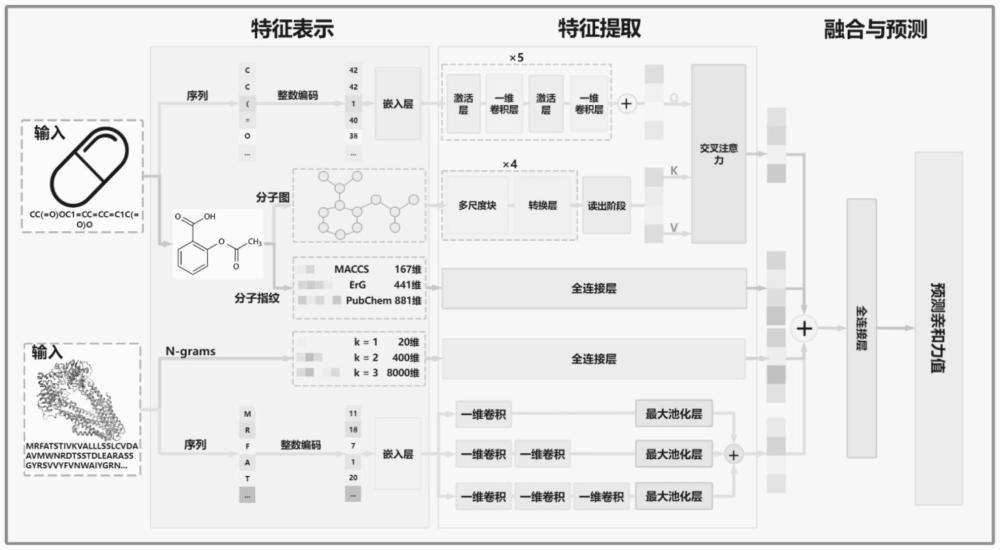
4、特征表示阶段从药物的smiles序列中提取出分子图和分子指纹特征,从靶标的蛋白质序列中提取出n-grams特征;
5、特征提取阶段的第一到第三通道分别用于提取药物的smiles序列、分子图和分子指纹特征,第四到第五通道分别用于提取靶标的蛋白质序列和n-grams特征;
6、特征融合阶段将药物和靶标提取到的特征进行特征融合连接起来并进行亲和力值的预测。
7、之所以需要进行亲和力预测,主要是为了以下作用:
8、虚拟筛选:在药物研发的早期阶段,利用药物-靶标亲和力预测方法,可以对大量化合物数据进行虚拟筛选,快速识别出具有潜在活性的化合物。这种方法可以大幅减少实验筛选的工作量和成本,提高筛选效率。
9、药物设计:在药物设计阶段,利用药物-靶标亲和力预测方法,可以帮助研究人员了解药物与靶标之间的相互作用机制,找到药物分子的最佳结合位点,指导药物分子的设计。这种方法有助于设计出更有效的药物分子,尽可能减少药物的副作用。
10、药物重定位(药物再利用):对于已知药物,利用药物-靶标亲和力预测方法,可以预测其与新靶标的结合亲和力,发现药物的新用途。这种方法可以为现有药物找到新的治疗领域,同时降低研发成本和时间。提高药物的经济效益和社会效益。
11、毒性评估:利用药物-靶标亲和力预测方法,预测药物与非靶标蛋白的结合亲和力,评估药物的潜在毒性。这种方法有助于在早期阶段识别和避免可能的副作用,提高药物的安全性。毒性评估是药物研发过程中的关键步骤,可以有效减少后期临床试验的失败风险。
12、当前药物研发面临诸多挑战,如疾病多样性和药物耐药问题凸显,而这些挑战又导致了研发周期长、成本高、成功率低等风险。通常情况下,从创新药物的研发到最后上市需要花费数十亿美元和10到15年的时间。研发一款新药所需的花费通常特别昂贵,并且十分耗时,常常还伴随着安全问题。
13、靶标是指在生物体内具有药效功能并能被药物作用,通过与药物分子相互作用来达到治疗疾病的目的的生物大分子。生物大分子如蛋白、核酸等是靶标的重要组成部分。识别新的药物与靶标之间的相互作用(drug-target interaction,dti)可以为药物研发提供关键信息,并且可以指导药物发现的过程,减少药物研发的时间和成本。具体来说,药物-靶标相互作用指的是细胞内的靶标(本文靶标指蛋白质)以适当的化学特性和亲和力与药物分子结合的过程,这是药物治疗疾病的内在机制之一。因此,选择新的和更合适的药物靶标以及在药物研发的早期阶段筛选出疗效良好的药物变得至关重要。预测药物与靶标之间的相互作用,对药物研发具有极其重要的意义。这一步骤能够帮助研究人员在药物研发的早期阶段,筛选出潜在的疗效良好的药物,同时也能够为研发人员提供有价值的信息,以便更好地选择新的、更合适的药物靶标。但是长期以来,药物-靶标相互作用的预测通常采用二元模型,只预测药物-靶蛋白间的相互作用,而忽视了药物-靶标间的亲和性。结合亲和力可以体现出药物与靶标对之间的相互作用强度,它在药物研发中发挥着非常关键的作用,如果结合强度不够强,则药物和靶标之间的相互作用可能是无用的。因此,研发预测药物和靶标之间结合亲和力的方法具有重要意义,而且更具挑战性。
14、传统的药物-靶标亲和力预测方法(drug-target binding affinity,dta)主要基于结构相似性,或基于生物信息学方法,如基于蛋白质序列或结构的对比方法。虽然这些方法一定程度上也可以预测药物-靶标的亲和力,但其仍存在许多局限性和缺陷。例如,基于结构相似性的方法容易受到配体和靶标的结构异质性的影响,导致预测结果不准确。而基于蛋白质序列或结构的对比方法则受限于数据量和质量,无法充分挖掘药物-靶标相互作用的复杂性和多样性。传统的药-靶亲和性预测方法一般仅能对药物-靶标间的相互作用进行预测,无法准确预测结合的强度,忽略了药物与靶标之间微小差异,从而导致预测结果的不确定性和误差较大。此外,传统方法需要大量实验数据支持,包括已知药物和靶标的亲和力、结构和性质等信息,但这些数据获取成本高昂,且不一定完整和准确,从而限制了其预测准确度和应用范围。同时,传统方法需要了解靶标的结构和功能,对于未知或未研究的靶标,其预测精度也会受到很大的限制。目前,传统的药物-靶标亲和力预测方法已不能满足临床需求,亟待发展新的方法和技术来解决这些问题。
15、随着人工智能技术的发展,深度学习是计算机视觉、语音识别和自然语言处理等多个领域的重要研究方向。最近几年,由于计算机硬件的能力持续提高以及数据的数量迅速增加,深度学习在医疗、药物研发等领域也逐渐得到应用,并取得了一定的成果。基于深度学习的方法已经在药物研发的各个领域中得到了广泛的应用。在药物分子的生成、优化和筛选方面,深度学习能够帮助设计出更加有效的药物分子。同时,深度学习还可以预测药物的毒性和副作用,为药物的安全性评估提供帮助。在药物治疗方面,深度学习可以应用于医学图像分析、医疗诊断和个性化治疗等方面,能够提高治疗效果并减少副作用。伴随着深度学习技术的出现,以此为基础的药物-靶标亲和力预测算法已逐步成为一个热门课题。深度学习方法通过多层次的非线性转换来提取数据的高级抽象特征,从而实现对大规模数据的有效建模。深度学习方法具有更高的预测精度和更广泛的适用性,通过深度学习算法对大量已知药物-靶标亲和力数据进行训练和学习,可以准确地预测新的药物-靶标亲和力。尤其是在大数据时代,基于深度学习的方法可以更好地利用海量的药物和蛋白质数据,并在计算效率和预测准确度上都有着很大的优势。与传统方法相比,深度学习可以学习到更复杂的模式和特征,同时还能够处理大量的数据,包括结构、生物信息学和药物活性数据等。深度学习方法的兴起也为药物-靶标亲和力预测提供了新的思路和方法。
16、因此为了解决上述问题,本发明拟采用基于深度学习的预测方法研究药物-靶标的结合亲和力值。相比于传统的药物-靶标亲和力预测方法,本发明所建立的模型可以更准确地捕捉药物和靶标之间的复杂关系,并提高预测精度。能够快捷地获得药物-靶标亲和力的预测结果,从而可以帮助药物研发人员在早期阶段发现适合药物的靶标,加速药物研发过程,节省时间和成本。有望为药物研发提供新的思路和方法,促进药物研发的进展。
17、优选地,从药物的smiles序列中提取出分子图和分子指纹特征的方法包括如下步骤:
18、我们构建了两个新的数据集db_k和db_e,除了使用上述两个新数据集外,还使用公共数据集davis上进行了训练和测试,数据来自于tefdta;
19、对于smiles序列特征表示的方法是先统计上述三个数据集中化合物的长度分布,然后定义一个固定长度,使得95%以上的化合物的长度小于上述长度;
20、对于分子图表示的方法是将smiles序列转换为相应的分子,再将分子转换为无向图,所述无向图上的每个节点都用22维向量表示;
21、对于分子指纹特征表示的方法是使用三种互补而全面的指纹,包括maccs指纹、pubchem指纹和药效团erg指纹,来捕获化合物的理化性质。
22、优选地,maccs指纹、pubchem指纹和药效团erg指纹三种指纹连接在以下公式中,以确保分子结构的精确表示,具体为:
23、fp=(fpmaccs||fppharmacophore erg||fppubchem)。
24、优选地,从靶标的蛋白质序列中提取出n-grams特征的方法包括如下步骤:
25、对于蛋白质smiles序列特征表示的方法是先统计上述三个数据集的蛋白质长度分布,然后定义一个固定长度,使得80%以上的蛋白质的长度小于上述长度;
26、对于蛋白质n-grams特征表示的方法包括:当n为1时,该特征枚举了20个标准氨基酸的出现频率,生成一个20维的特征向量,当n设为2时,列举每个可能的二肽序列的频率,生成一个400维的特征向量,随着n值的增加,按照上述增加模式,最后将n个特征向量连接起来以表示蛋白质。
27、优选地,当n分别设置为1,2,3时,为每个蛋白质序列获得一个8420维,即201+202+203的的特征向量,归一化处理表示如下:
28、
29、其中,xn表示n-grams特征向量,均值xn表示xn的平均值,std(xn)表示xn的标准差。
30、优选地,五个通道中,第一与第五通道分别使用残差扩张卷积神经网络和多尺度卷积神经网络提取药物smiles序列和靶标的蛋白质序列特征;
31、第二个通道采用多尺度图卷积神经网络提取分子图的特征;
32、第三与第四通道使用全连接神经网络提取分子指纹和n-grams特征。
33、优选地,还包括有应用交叉注意块来捕获药物的smiles序列和分子图之间的交互特征的步骤,具体包括:
34、ds是用于提取smiles序列特征的残差扩张卷积神经网络模块的输出,dg是用于提取分子图特征的多尺度图卷积神经网络模块的输出;查询(q)、键(k)、值(v)计算如下:
35、q=fq(ds)=dswq
36、k=fk(dg)=dgwk
37、v=fv(dg)=dgwv
38、其中q是基于ds的投影生成的查询,k和v分别是基于ds和dg的投影生成的键和值;
39、wq、wk、wv是在训练过程中自动学习的权重矩阵,然后对q、k、v矩阵进行交叉注意运算,公式为:
40、
41、drug_equence_graph_feature=crossattention(q,k,v)=attention_map*v
42、其中,交叉注意力模块的头数h=1,嵌入维度d=96。
43、本发明除了提供了一种基于多层次特征融合的药物-靶标亲和力预测方法,还提供了一种药物-靶标亲和力预测系统,包括:
44、特征表示模块:从药物的smiles序列中提取出分子图和分子指纹特征,从靶标的蛋白质序列中提取出n-grams特征;
45、特征提取模块:第一到第三通道分别用于提取药物的smiles序列、分子图和分子指纹特征,第四到第五通道分别用于提取靶标的蛋白质序列和蛋白质n-grams特征;
46、特征融合与预测模块:将药物和靶标提取到的特征进行特征融合连接起来并进行亲和力值的预测。
47、总之,本发明的方案具有如下有益效果:
48、(1)使用最新版本的bindingdb数据库来收集和归档数据,构建得到一个新的数据集,称为db_k,有34279个kd值的样本,与之前的工作相比增加了大约20000个样本。此外,还获得了另一个新的数据集,称为db_e,包含105936个带有ec50值的样本。
49、(2)本发明提出了一种基于多层次特征融合的dta预测方法,称为mlff-dta。mlff-dta以药物的smiles序列和靶标的蛋白质序列作为输入。从药物的smiles序列中提取出分子图和分子指纹特征,从蛋白质序列中提取出n-grams特征。从不同的角度表征化合物和蛋白质,更好地特征表示。
50、(3)目前,药物分子(化合物)表征可分为smiles序列、图和指纹三大类。可以从smiles序列中提取出化合物的序列特征,从分子图中提取出结构构型特征,从分子指纹中提取化合物的理化性质特征。因此,本发明使用这三种类型的特征来捕获化合物的多层次信息,用于dta预测任务。
51、(4)maccs指纹记录是否存在预定义的官能团、子结构和片段。pubchem指纹包含七个信息部分,如单原子对、单原子的最近邻和化学环等。药效团erg指纹图谱记录了两个药效团点之间的距离,更详细地描述了分子结构信息。这三种指纹从不同的角度描述了化合物的物理和化学性质。本发明只用这三种互补的指纹融合作为一种新的指纹特征,称为fp,来描述化合物的物理和化学性质,用于预测dta。
52、(5)在k折交叉验证过程中,可以生成k个模型,用于这些k个模型的训练数据集并不完全相同,这导致训练模型的权重也不同。将交叉验证过程中获得的模型组合起来,可以生成具有更高预测精度的大模型。这种方法在不增加训练资源消耗的情况下有效地提高了预测精度,只是在计算过程中占用了适当的内存。
技术研发人员:王娟,王姣,幸佳杰,张媛,韩伟亮
技术所有人:内蒙古大学
备 注:该技术已申请专利,仅供学习研究,如用于商业用途,请联系技术所有人。
声 明 :此信息收集于网络,如果你是此专利的发明人不想本网站收录此信息请联系我们,我们会在第一时间删除