学习系统、确定系统和预测系统以及学习方法、确定方法和预测方法与流程
本发明涉及一种测定生物标记的值的技术。
背景技术:
1、在dna(deoxyribonucleic acid:脱氧核糖核酸)中,已知发生被称为“甲基化”的现象。甲基化是指基于甲基分子与胞嘧啶化学键合的修饰。该胞嘧啶(c:cytosine)与鸟嘌呤(g:guanine)、腺嘌呤(a:adenine)、胸腺嘧啶(t:thymine)一起构成了构成dna的4个必需核酸碱基。核酸碱基的任意序列被称为“核苷酸序列”,对蛋白质等的重要信息进行编码的核苷酸序列被称为“基因组序列”或“基因”。
2、在人体中,在dna链上胞嘧啶紧与鸟嘌呤连接的位置(称为“cpg位点”)上,甲基化尤其常见。甲基化状态影响基因的激活或抑制化,某些基因的cpg位点的甲基化状态形成许多疾病的重要的生物标记。通常,为了制作疾病诊断的定量模型,使用从几个生物标记候选序列的组合获得的数据。因此,测定生物标记的dna甲基化变得重要。
3、在dna的测定过程中,数据出现错误,对任何推测/预测的可靠性都会带来影响。用于将生物标记的选择最佳化的以往的研究在测定工艺中假设仅有极少的错误,仅将焦点集中在可利用的数据的预测值上。作为这种方法的例子,已知有一种特征选择算法,其依赖于来自(artificial intelligence(人工智能)分类器的性能之类的)定量模型的输出信号来确定是否使用生物标记序列作为用于分类的特征。
4、关于这种以往的技术,例如在专利文献1中记载有从代表性的生物标记数据选择生物标记集进行评价的内容。并且,在非专利文献1中记载有pcr偏差(pcr:polymerasechain reaction(聚合酶链式反应))的测定及松弛。
5、以往技术文献
6、专利文献1:日本特表2017-523437号公报
7、非专利文献
8、非专利文献1:“measuring and mitigating pcr bias in microbiome data”、justin d.silverman等、[2022年3月22日检索]、互联网(https://www.biorxiv.org/content/10.1101/604025v1)
技术实现思路
1、发明要解决的技术课题
2、在下一部分中,对所要学习生物标记序列(sequence:序列)的测定误差特性的现有研究进行详细讨论。对这些现有研究和与它们相关的问题进行讨论,并进行各个阶段的详细说明。
3、[dna的甲基化测定]
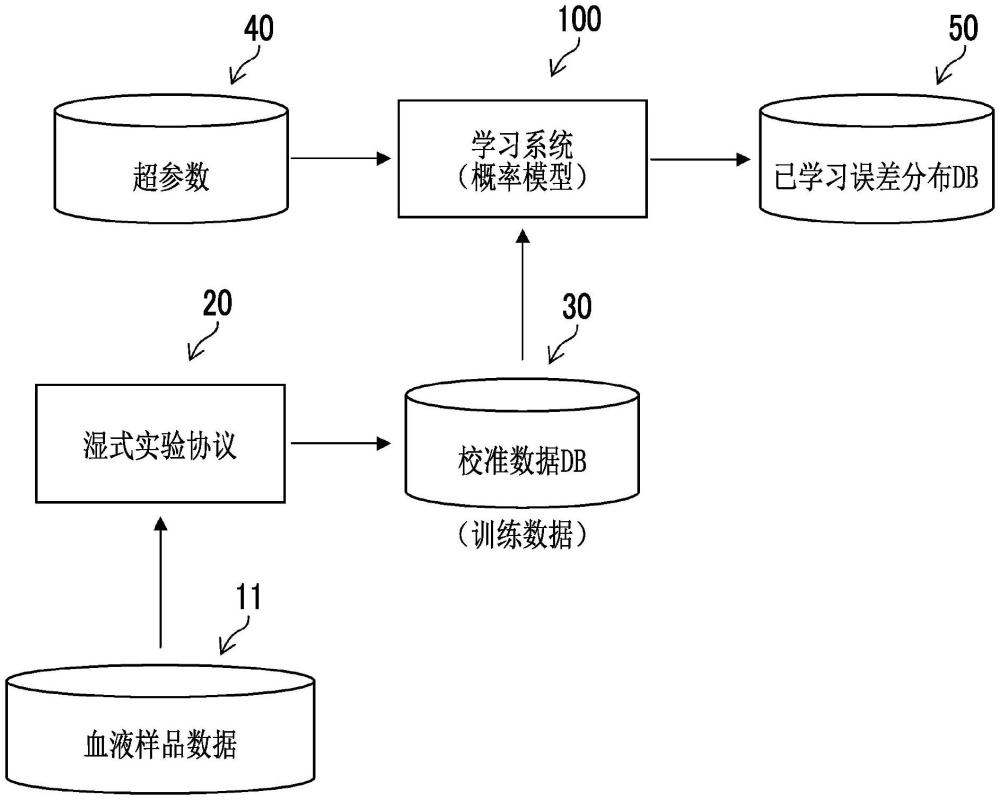
4、将甲基化测定的概要示于图1中。在甲基化的测定中,血液样品10被亚硫酸氢盐转化,通过pcr装置扩增基因/信号,并通过新时代测序仪等进行测定。这些一系列的测定顺序构成湿式实验协议20(wet experiment protocol)。
5、[step1:亚硫酸氢盐转化]
6、为了区分cm(甲基化胞嘧啶)和cu(非甲基化胞嘧啶),使用亚硫酸氢盐转化(bisulfite conversion)的追加步骤。在亚硫酸氢盐转化中,cu转化为尿嘧啶(u:uracil),cm仍为cm。若所转化的样品被序列化,则cm作为c(胞嘧啶)而读出,另一方面,尿嘧啶作为胸腺嘧啶而读出。由此,能够区分胞嘧啶的甲基化状态。
7、[问题1:亚硫酸氢盐转化中的问题]
8、该顺序的理想结果为cu被转化为100%的尿嘧啶、cm完全不会转化为尿嘧啶(转化为0%,cm仍为cm)。但是,在化学反应的性质上,转化的成功(或不成功)的程度是概率论的,定量研究是困难的。以下将这种亚硫酸氢盐转化的不完全性称为“问题1”。
9、[step2:pcr扩增]
10、该阶段可以理解为测定的信号扩增阶段。标准上(即,不是为了进行甲基化而是为了进行亚硫酸氢盐转化),各个“信号”是感兴趣的基因或序列。在原始数据中,这种序列的数量非常少,因此所派生的信号较弱。因此,认为通过多次复制原始序列,能够增加序列数而扩增信号。例如,将pcr前的基因1的信号强度称为g1_pre,将pcr后的信号强度称为g1_post。另外,实际上,将焦点集中在同时扩增多个基因/信号上。因此,关于基因2,以与基因1相同的方式定义g2_pre和g2_post。
11、现在,若首先进行上述step1,则即使是仅1个基因,也可获得2个信号。例如,基因1具有几个被转化为包含尿嘧啶的其他序列的、作为非甲基化具有cpg的序列。同样地,cpg被甲基化的序列不会被转化。这是常见的,在肝脏和胃的dna的混合物中被发现。在这种混合物中,有可能对肝脏重要的基因在肝细胞中未被甲基化,但在胃细胞中被甲基化(因此被抑制)。因此,关于基因1,将pcr前信号的强度和pcr后信号的强度设为g1_u_pre及g1_u_post(在未被甲基化的情况)、设为g1_m_pre及g1_m_post(在被甲基化的情况),将所解密的序列设为g1_m_pre及g1_m_post。
12、[问题2:单亚硫酸氢盐协议中的pcr偏差]
13、即使扩增相同基因的信号,亚硫酸氢盐转化也会成为2个信号类型。因此,g1_u_post/g1_u_pre=g1_m_post/g1_m_pre不成立。已知即使在g1_u_pre=g1_m_pre的情况下,扩增后成为g1_u_post/g1_u_pre>g1_m_post/g1_m_pre(即,非甲基化基因相对于甲基化基因过度扩增)。但是,这种非甲基化信号的过度扩增的程度依赖于基因序列本身和测定中使用的化学物质。以下,将该问题称为“问题2”。
14、[问题3:pcr扩增中的问题]
15、pcr的理想结果为g1_post/g1_pre=g2_post/g2_pre。但是,实际上,某种基因序列比其他基因序列容易测定而该等价性不成立。将这种在一起扩增了几个不同的生物标记序列/基因的情况下产生的偏差称为多重化协议中的“pcr偏差”(以下,称为“问题3”)。
16、[以往技术中的对应]
17、对上述的问题1~3的以往技术中的对应进行说明。在以往技术中,关于问题1,定量研究中通常不需要极端的准确度,因此未考虑亚硫酸氢盐转化的成功程度。并且,关于问题3,在迄今为止的微生物学的研究中,以乘法的方式考虑pcr的效果。即,在以往的技术中,认为若1次pcr循环后的基因1的信号强度为j,则2次循环后的信号强度为j2,x次循环后的信号强度同样为jx。使用该假设,pcr被模型化为使用了多项逻辑-通常线性模型的对数线性过程。“批次效应”(针对每个批次显示出稍微不同的偏差特性的样本的pcr)等其他协变量也包含在概率论方法中。模型在“训练”所生成的校准数据之后,用于校准pcr偏差。
18、并且,关于问题2,单一协议设定中的测定误差和偏差的特征化更简单,因此在一部分的pcr数据中,为了发现偏差的程度而进行线性回归。在计算出线性回归估计量之后,能够使用该方程式来校准这种偏差。
19、dna甲基化的准确测定的重要性已经叙述。在如疾病诊断的应用领域中,使用多个生物标记序列的数据并输入到定量模型中并不少见。在设计同时测定多个生物标记的指标值的测定工艺时,问题1、问题2、问题3被组合在一起,这些全部成为问题。因此,误差的定量化和学习误差的特性变得非常困难。在本发明中,对在存在3个问题的情况下学习测定误差特性并且使所学习的误差特性反映到生物标记选择基准的系统进行研究。在存在该问题的组合的情况下,解决针对dna的甲基化的测定误差特性评价的问题,形成本发明的主要新颖性。
20、本发明在如液体生命学(liquid biopsy)那样需要同时且非常准确地测定来自多个基因的dna甲基化的情况下尤为重要。尤其,已知为了准确鉴定如癌症的疾病,某种癌细胞基因与健康细胞中的相同基因相比显示出高的甲基化。在这种情况下,问题2是指测定时从癌症和正常的dna的混合低估了真实的甲基化比(负的偏差)。问题1和问题3进一步使低估的程度恶化。
21、本发明是鉴于上述情况而完成的,其一方式提供一种学习生物标记序列的测定误差特性的学习系统及学习方法。并且,本发明的一方式提供一种反映所学习的误差特性来确定序列集的确定系统和确定方法、以及使用通过学习系统或学习方法获得的数据来预测基因序列的测定误差特性的预测系统和预测方法。
22、用于解决技术课题的手段
23、本发明的第1方式所涉及的学习系统为学习测定协议变量与作为生物标记序列的结果而产生的误差特性的关系的学习系统,其具备处理器,处理器进行如下处理:输入以能够针对具有重要性的变量获取适当的数据的方式设计的校准数据;及使用概率模型,学习针对具有重要性的变量遍及各测定协议的误差分布的特性,概率模型包括:第1参数,为了对亚硫酸氢盐转化的误差进行模型化,用适当选择的先验参数进行了初始化;第2参数,为了对生物标记序列的扩增的相互依赖性进行模型化,用适当选择的先验参数进行了初始化;及第3参数,为了对pcr整体的偏差进行模型化,用适当选择的先验参数进行了初始化。第1方式所涉及的学习系统为学习测定协议变量与作为生物标记序列的结果而产生的误差特性之间的关系(被定义为模板对产物比)的系统。
24、在第1方式及以下各方式中,“具有重要性的变量”为由实验室的专家已知对信号扩增性能带来影响的变量,对于这种变量,pcr装置进行调整。例如,如后述的图2所示的pcr温度或pcr循环数为“具有重要性的变量”的一例。若温度过高,则dna被分解,不会发生为了复制目标基因序列而需要的反应。并且,关于第1~第3参数,可以使用相同的参数作为“适当地选择的先验参数”。并且,关于“校准数据(calibration data)的输入”,例如在pcr温度的情况下,要求能够在通常的pcr中使用的温度的范围内适当的显示。
25、第2方式所涉及的学习系统在第1方式中,第2参数为如下参数:分别获取亚硫酸氢盐转化后的基因的甲基化序列及非甲基化序列的计数,针对甲基化序列及非甲基化序列的各序列,将所获取的计数以能够分别确定先验变量的多项分布进行了模型化。第2方式规定用于对应于上述的问题2的第2参数的具体方式,将亚硫酸氢盐转化的误差进行模型化及修正,以使能够正确地评价生物标记序列的甲基化。在第2方式中,能够根据经验性数据分析来选择更优异的先验变量。另外,在第2方式中获取的计数能够根据如碱基序列的gc比(鸟嘌呤与胞嘧啶之比)的因素来进行模型化。
26、第3方式所涉及的学习系统在第1或第2方式中,第3参数为如下参数:在使用通用引物同时扩增多个序列的情况下,施加了以多项分布计算的计数的各个计数的合计遵循高斯分布这一结构数据限制。第3方式规定用于对应于上述的问题3的第2参数的具体方式,在多个生物标记的数量多且以能够计算结构数据限制的方式简化模型化参数的情况下,多个分散计数中的各计数的合计遵循高斯分布。并且,在同时扩增多个序列的情况下,进行如各标记的计数值并非独立且合计值成为大致恒定的扩增方式,因此适合如上所述的基于多项分布的模型化。此外,在伴随亚硫酸氢盐转化的甲基化测量中,各标记物具有甲基化、非甲基化这2种状态,因此成为相对于标记数×2的计数值的模型化。
27、本发明的第4方式所涉及的确定系统为具备处理器的确定系统,处理器进行如下处理:输入在多重化面板中使用的关注生物标记序列的核苷酸序列和测定协议信息;从第1至第3方式中任一项所涉及的学习系统输入所学习的误差特性和与误差特性建立关联的元数据;使用利用预先确定的基准进行输入的核苷酸序列、测定协议信息、所学习的误差特性和元数据来输出用于集合可能的生物标记序列的第1分数;及考虑针对各集合的第1分数的值来确定生物标记序列集。在第4方式所涉及的确定系统中,为了确定在多重面板中是否使用生物标记序列,使用来自第1方式所涉及的系统的输出。第1分数为源自测定精度的分数,并且为测定误差越小值越高的“低误差分数”。
28、第5方式所涉及的确定系统在第4方式中,处理器进行如下处理:针对每个应确定的生物标记序列输入第2分数;及考虑针对生物标记序列集中的各生物标记序列的第1分数,将第1分数与第2分数的平衡最佳化,由此选择多重化面板的最佳子集。在第5方式所涉及的确定系统中,通过考虑多重化面板(multiplex panel)的最终目标,为了能够进行生物标记序列的更加平衡的取舍的选择,增强第4方式。例如与所要预测的疾病的相关性越大,第2分数为越高的分数(相关性分数)。并且,例如通过算出由第1分数与第2分数的相加平均或相乘平均规定的第3分数并且将该第3分数最大化,能够使“第1分数与第2分数的平衡”最佳化。
29、本发明的第6方式所涉及的预测系统为预测基因序列的测定误差特性的预测系统并且具备处理器,处理器进行如下处理:输入在多重化面板中使用的关注生物标记序列的核苷酸序列和测定协议信息;从第1至第3方式中任一项所涉及的学习系统输入所学习的误差特性和与误差特性建立关联的元数据;使用用于计算2个基因序列之间的相似性的尺度的测定基准来计算以前包含在校准数据中的生物标记序列与新的生物标记序列的相似度;及将计算出的相似度与其他相关的输入和所学习的误差特性组合使用而预测测定不包含在校准数据中的生物标记序列时的误差特性。第6方式所涉及的预测系统能够将第1~第3方式所涉及的学习系统用于不包含在校准数据中的生物标记序列。
30、另外,在第6方式中,“其他相关的输入”例如是指与生物标记序列对应的元数据。例如,若基因类型为“启动子或增强子”、cpg类型为“岛屿、岛岸、岛架”且cg的丰富程度为“高、低”,则针对某一生物标记序列g1的这些信息的组合(元数据的一例)能够表示为“启动子、岛屿、低”这一矢量。
31、第7方式所涉及的预测系统在第6方式中,处理器进行如下处理:使用所预测的误差特性来获取与不包含在校准数据中的生物标记序列最相似的、能够在校准数据中利用的生物标记序列;及将所获取的生物标记序列的信息反映到第4或第5方式所涉及的确定系统中的生物标记序列集的确定中。在第7方式中,使用第4或第5方式所涉及的确定系统,在生物标记序列集选择中,能够使用不包含在校准数据中的生物标记序列。
32、本发明的第8方式所涉及的学习方法为由具备处理器且学习测定协议变量与作为生物标记序列的结果而产生的误差特性的关系的学习系统执行的学习方法,其中,处理器进行如下处理:输入以能够针对具有重要性的变量获取适当的数据的方式设计的校准数据(校准数据输入步骤);及
33、使用概率模型,学习针对具有重要性的变量遍及各测定协议的误差分布的特性(学习步骤),概率模型包括:第1参数,为了对亚硫酸氢盐转化的误差进行模型化,用适当选择的先验参数进行了初始化;第2参数,为了对生物标记序列的扩增的相互依赖性进行模型化,用适当选择的先验参数进行了初始化;及第3参数,为了对pcr整体的偏差进行模型化,用适当选择的先验参数进行了初始化。第8方式规定与上述的第1方式对应的学习方法。
34、第9方式所涉及的学习方法在第8方式中,第2参数为如下参数:分别获取亚硫酸氢盐转化后的基因的甲基化序列及非甲基化序列的计数,针对甲基化序列及非甲基化序列的各序列,将所获取的计数以能够分别确定先验变量的多项分布进行了模型化。第9方式规定与上述的第2方式对应的学习方法。
35、第10方式所涉及的学习方法在第8或第9方式中,第3参数为如下参数:在使用通用引物同时扩增多个序列的情况下,施加了以多项分布计算的计数的各个计数的合计遵循高斯分布这一结构数据限制。第10方式规定与上述的第3方式对应的学习方法。
36、本发明的第11方式所涉及的确定方法为由具备处理器的确定系统执行的确定方法,其中,处理器进行如下处理:输入在多重化面板中使用的关注生物标记序列的核苷酸序列和测定协议信息(序列信息输入步骤);输入作为第8至第10方式中任一项所涉及的学习方法的结果获得的、所学习的误差特性和与误差特性建立关联的元数据(学习结果输入步骤);使用利用预先确定的基准进行输入的核苷酸序列、测定协议信息、所学习的误差特性和元数据来输出用于集合可能的生物标记序列的第1分数(分数输出步骤);及考虑针对各集合的第1分数的值来确定生物标记序列集(序列集确定步骤)。第11方式规定与上述的第4方式对应的确定方法。
37、第12方式所涉及的确定方法在第11方式中,处理器进行如下处理:针对每个应确定的生物标记序列输入第2分数(分数输入步骤);及考虑针对生物标记序列集中的各生物标记序列的第1分数,将第1分数与第2分数的平衡最佳化,由此选择多重化面板的最佳子集(子集选择步骤)。第12方式规定与上述的第5方式对应的确定方法。
38、本发明的第13方式所涉及的预测方法为由具备处理器且预测基因序列的测定误差特性的预测系统执行的预测方法,其中,处理器进行如下处理:输入在多重化面板中使用的关注生物标记序列的核苷酸序列和测定协议信息(序列信息输入步骤);输入通过第8至第10方式中任一项所涉及的学习方法获得的、所学习的误差特性和与误差特性建立关联的元数据(学习结果输入步骤);使用用于计算2个基因序列之间的相似性的尺度的测定基准来计算以前包含在校准数据中的生物标记序列与新的生物标记序列的相似度(相似度计算步骤);及将计算出的相似度与其他相关的输入和所学习的误差特性组合使用而预测测定不包含在校准数据中的生物标记序列时的误差特性(误差特性预测步骤)。第13方式规定与上述的第6方式对应的预测方法。
39、第14方式所涉及的预测方法在第13方式中,处理器进行如下处理:使用所预测的误差特性来获取与不包含在校准数据中的生物标记序列最相似的、能够在校准数据中利用的生物标记序列(序列获取步骤);及将所获取的生物标记序列的信息反映到第11或第12方式所涉及的确定方法中的生物标记序列集的确定中(信息反映步骤)。第14方式规定与上述的第7方式对应的预测方法。
40、另外,由处理器执行上述的方式的学习方法、确定方法和预测方法的程序(学习程序、确定程序、预测程序)和记录有这些程序的计算机可读代码的非暂时性记录介质也包括在本发明的范围内。
41、发明效果
42、如以上说明,本发明所涉及的学习系统、确定系统和预测系统以及学习方法、确定方法和预测方法具有以下效果。
43、(1)能够处理一起测定多个基因序列而多重化的面板。
44、(2)能够处理经亚硫酸氢盐转化的样品。
45、(3)能够使用序列参数和协议参数作为输入来预测测定误差。
46、(4)能够确定是否将序列用于分析/分类的目的。
技术研发人员:J·辛格
技术所有人:富士胶片株式会社
备 注:该技术已申请专利,仅供学习研究,如用于商业用途,请联系技术所有人。
声 明 :此信息收集于网络,如果你是此专利的发明人不想本网站收录此信息请联系我们,我们会在第一时间删除