用于分子虚拟筛选的集成回归模型及生成方法、预测方法与流程
本发明涉及计算机辅助药物设计,尤其涉及一种用于分子虚拟筛选的集成回归模型及生成方法、预测方法。
背景技术:
1、虚拟筛选技术尽管能够覆盖数百万种化合物,但相较于已知的类药物分子结构总数(约1063),其覆盖范围仍相对狭窄。因此,如何在药物发现的早期阶段大幅度扩展可探索的分子数量,已成为当前药物研发领域亟待解决的问题。
2、随着按需合成的化合物库的出现,用于虚拟筛选的可购买化合物的数量近年来呈爆炸式增长,已超过了10亿种化合物。尽管目前已有不少研究成功应用超过10亿种化合物的超大规模分子库进行虚拟筛选,但是确实存在一些局限性。首先,筛选大规模数据库将不可避免地需要大量的计算资源。例如,基于配体的虚拟筛选,为包含1010个分子的数据库生成构象集的成本约为20000美元。针对热休克蛋白90(hsp90)的基于结构的虚拟筛选,在使用了45000个cpu的情况下,总计耗时18小时,而单个cpu需要约55年。这项成本为15000美元,且该费用仅用于搜索数据库,而不包括其生成或存储的成本。
3、另一方面,对于没有足够数量已知活性配体的靶点,无法采用基于配体的虚拟筛选方法快速过滤大型虚拟库,只能采用基于结构的筛选方法。当前已有研究采用基于结构的药物设计工作流程筛选超大规模数据库。这些研究需要大量的计算资源,然而这些资源对许多学术研究人员来说是无法获得的。此外,这种高昂的计算成本使得将这种策略应用于许多不同的蛋白质靶标变得不切实际。随着虚拟库变得越来越庞大,必须制定新的策略来缓解这些详尽筛选活动的计算成本。以在精度损失最小的情况下,在数十亿分子库中找到最佳评分的配体。
4、为应对这一问题,引入了主动学习(active learning,al)方法。主动学习方法不仅拓宽了超大规模虚拟筛选的应用范围,还为实验室条件下快速实现超大规模虚拟筛选提供了可行的解决方案。主动学习作为一种无需额外资源即可提升筛选速度的方法,其核心在于使用统计模型替代对接算法,避免进行耗时的蛮力搜索。al算法在药物发现领域的应用可追溯到warmuth等人的开创性工作,他们的研究突显了al在处理药物研发中的标记任务方面的潜力。在过去的二十年中,al在多个药物开发项目中展现出广阔的应用前景,包括聚焦库设计、理性的de novo设计、药物组合、跟踪细胞、分子性质预测以及活性化合物的寻找等。
5、先前的研究主要集中在利用al算法识别理想样本,并通过额外的训练数据提升机器学习(ml)模型的能力。根据不同的应用需求,可以通过灵活调整采样方法来改变al的偏好。例如,探索导向的采样方法倾向于选择具有新颖结构的样本,以拓宽模型的适用领域。而开发导向的采样方法则更注重选择具有高性能的样本,如针对特定靶点的化合物结合亲和力,这种方法通常能显著提高命中率。采样方法,即查询策略,是主动学习方法成功的关键。不恰当的采样方法可能导致模型预测性能不佳,进而需要更多的主动学习迭代。
技术实现思路
1、本发明旨在解决不恰当的采样方法导致模型预测性能不佳的问题。为此,本发明提供一种用于分子虚拟筛选的集成回归模型及生成方法、预测方法,其核心技术原理在于利用主动学习策略和多种回归模型的集成来提高虚拟筛选的效率和准确性。本发明通过在得分最高的化合物中选择预测值标准差最大的化合物进行标注,以将精力集中于那些最具挑战性或者最不确定的样本,从而提高标注效率和模型性能。同时,充分利用不同模型之间的差异性和多样性,通过加权平均的方式将各模型之间的预测值结合起来作为最终预测结果,以降低单个模型的偏差,并提高整体模型的性能和鲁棒性。本发明特别适用于超过10亿种化合物的超大规模化合物库。
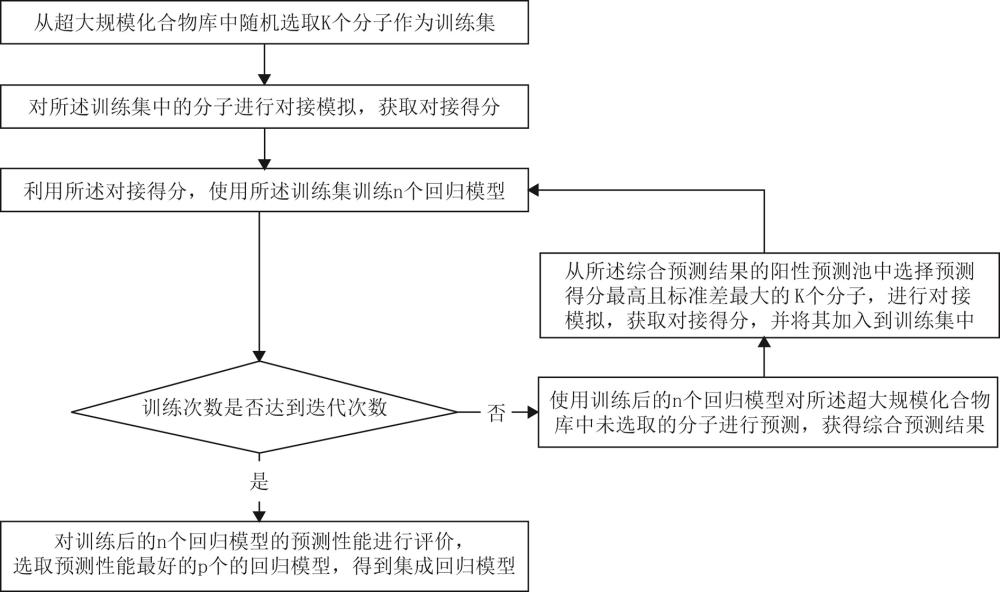
2、本发明提供一种用于分子虚拟筛选的集成回归模型生成方法,采用的技术方案如下:包括以下步骤:
3、步骤1:从超大规模化合物库中随机选取k个分子作为训练集;
4、步骤2:对所述训练集中的分子进行对接模拟,获取对接得分;
5、步骤3:利用所述对接得分,使用所述训练集训练n个回归模型;
6、步骤4:判断训练次数是否达到迭代次数,若未达到迭代次数,进入步骤5,若达到迭代次数,进入步骤7,
7、步骤5:使用训练后的n个回归模型对所述超大规模化合物库中未被选取的分子进行预测,获得综合预测结果;
8、步骤6:从所述综合预测结果的阳性预测池中选择预测得分最高且标准差最大的k个分子,进行对接模拟,获取对接得分,并将其加入到训练集中;进入步骤3;
9、步骤7:对训练后的n个回归模型的预测性能进行评价,选取预测性能最好的p个的回归模型,得到集成回归模型,n>p。
10、进一步的,对从超大规模化合物库中选取的分子进行处理,所述处理包括确定分子的立体异构构型和正确的电离态,以及利用rdkit将smiles格式的分子转换为ecfp4指纹。
11、进一步的,在步骤5中,以n个回归模型的预测结果的加权平均值作为综合预测结果。
12、进一步的,所述k为超大规模化合物库中分子总数的1%。
13、进一步的,所述迭代次数为8次。
14、进一步的,在步骤6中,选择预测得分最高且标准差最大的k个分子的过程为:
15、根据预测得分对所有分子进行排序,选择得分最高的n个分子,n>k;
16、计算n个分子中每个分子的预测值的标准差,选择标准差最大的k个分子。
17、进一步的,所述回归模型包括rf(随机森林)、svm(支持向量机)、ridge(岭回归)、xgboost(极限梯度提升)、lgbm(轻量梯度提升)、dnn(深度神经网络)、gcn(图卷积网络)和gat(图注意力网络)中的多个。
18、进一步的,在步骤7中,以rmse作为回归模型的预测性能的评价指标。
19、本发明还提供一种用于分子虚拟筛选的集成回归模型,采用的技术方案如下:通过上述的一种用于分子虚拟筛选的集成回归模型生成方法从8个回归模型中选取4个回归模型作为集成回归模型;
20、回归模型包括:rf、svm、ridge、xgboost、lgbm、dnn、gcn和gat;
21、集成回归模型由svm、xgboost、gcn和gat组成。
22、本发明还提供一种用于分子虚拟筛选的预测方法,采用的技术方案如下:使用上述的一种用于分子虚拟筛选的集成回归模型,包括以下步骤:
23、s1:获取待对接分子;
24、s2:将所述待对接分子的ecfp4指纹分别输入svm和xgboost,计算得到svm预测结果和xgboost预测结果;将所述待对接分子的图的分子表征分别输入gcn和gat,计算得到gcn预测结果和gat预测结果;
25、s3:计算svm预测结果、xgboost预测结果、gcn预测结果和gat预测结果的加权平均值,得到最终预测结果。
26、本发明实施例中的上述一个或多个技术方案,至少具有如下技术效果之一:
27、1、计算效率显著提升。在计算用时方面,本发明在使用主频为2.25ghz的amd epyc7742 cpu(128核心)以及1块nvidia gtx2080 gpu的情况下,模型训练和筛选10亿分子库共用时950个小时。相较于传统的分子对接方法,如glide,效率提升了180倍。本发明的高效的计算能力显著减少了筛选时间,为大规模虚拟筛选提供了切实可行的解决方案。
28、2、资源利用率优化。本发明通过集成多种回归算法并结合预测得分最高和标准差最大的采样策略,有效利用了不同模型之间的差异性和多样性,提高了整体模型的性能和鲁棒性。通过智能选择最具挑战性的样本进行标注,减少了不必要的计算资源浪费,从而在保持高精度的同时,大幅降低了计算成本和时间投入。
29、3、筛选效果卓越。通过采用本发明的集成回归模型(adel模型),从超过10亿个化合物的虚拟分子库中筛选出38个候选化合物,随后通过湿实验进行了验证。在筛选出的38个化合物中,有10个化合物展示了强效的生物活性,特别是两个化合物的ic50值小于100nm。这些化合物有望为β2ar相关疾病(如哮喘和慢性阻塞性肺病)的治疗提供新的候选药物,展示了adel模型在药物发现领域的巨大潜力。adel模型显著提高了药物发现过程中的效率和准确性,通过减少虚拟筛选的计算时间和资源消耗,研究人员可以更快地识别和验证潜在的药物候选化合物,加速药物研发的进程。
30、本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
技术研发人员:周晓菲,王目阔
技术所有人:天津贝芸科技有限公司
备 注:该技术已申请专利,仅供学习研究,如用于商业用途,请联系技术所有人。
声 明 :此信息收集于网络,如果你是此专利的发明人不想本网站收录此信息请联系我们,我们会在第一时间删除